金属−氢(M−H)相互作用在各种能源及小分子转化反应过程中广泛存在。其中,金属钯(Pd)是相关体系中最具代表性的催化材料。在诸多钯催化的加氢反应中,电催化二氧化碳还原反应(CO2RR)可将温室气体CO2转化为高价值化学品(如HCOOH等),作为实现我国“双碳”战略的有效途径,受到广泛关注。当前对于Pd催化性能的提高主要是通过合金元素(Au或Ag)掺杂调控Pd电子结构,从而调控反应中间体的吸附能。最新研究表明,Pd在电催化加氢过程中往往形成氢化物(PdHx),而氢原子同样被视作一种合金元素进一步调控Pd催化位点电子结构及中间体的吸附。然而,在工况条件下,Pd的原位氢化过程及其影响因素的精细表征(尤其是定量测量)较为困难,各种常规表征方法在灵敏度和时间分辨率等方面具有一定的局限性,限制了相关催化机制的阐明。为了更深入理解催化表界面处的电催化材料、电催化微环境和反应物分子之间的关系,有必要发展新的原位表征方法(最好是基于不同的测试原理)进行进一步的精细表征。
近日,威尼斯wnsr888(亚洲)集团有限公司丁梦宁课题组在前期发展的原位电输运谱(Electrical transport spectroscopy, ETS)测量表征技术基础上,和苏州大学功能纳米与软物质研究院李彦光课题组合作,实现了钯基金属催化材料体系在CO2RR过程中H动力学的原位定量表征,揭示了电催化表界面处局域pH,质子供体pKa和CO2分子对相变过程的影响及其与CO2RR的关联。该成果以“Critical role of hydrogen sorption kinetics in electrocatalytic CO2 reduction revealed by on-chip in situ transport investigations”为题发表于Nature Communications。我院2020级博士生穆张岩和苏州大学功能纳米与软物质研究院副教授韩娜为论文的共同第一作者。
电输运谱(ETS)是一种基于微纳电化学器件集成平台的原位输运测量表征方法,近年来已被应用于多种电化学体系的原位表界面过程研究,包括金属催化材料表面吸附(Nat. Commun.2015, 6: 7867; ACS Cent. Sci. 2018, 4: 590; J. Phys. Chem. Lett. 2020, 11: 5798)、金属氧化物/羟基氧化物原位相变(Angew. Chem.2021, 133, 16584; J. Am. Chem. Soc.2022, 144, 15185)、二维半导体催化材料的“自门控”效应(Nat. Mater.2019, 18: 1098)及单原子催化位点处的“位点静电极化”作用(Nat. Commun. 2022, 13: 3063)等。本工作主要选取了Pd和Pd4Ag为模型催化剂,系统研究了钯基金属在CO2RR过程中的H动力学及相变过程,选取铂(Pt)为典型的表面H吸附材料进行对比。

图1 原位电输运测试电化学池和原理示意图
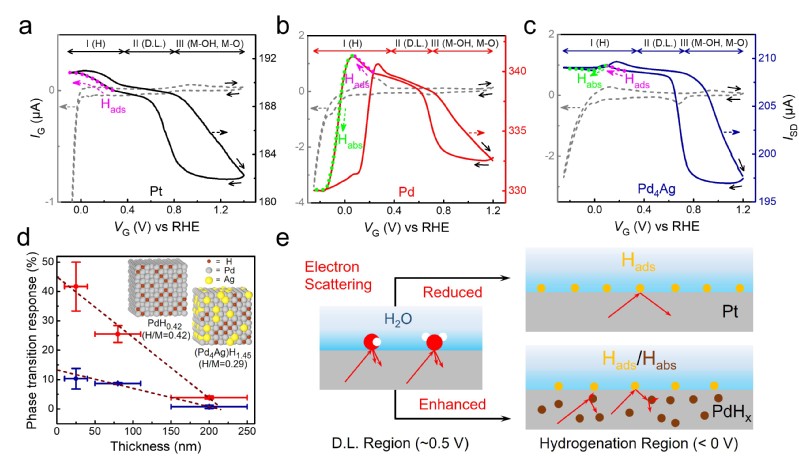
图2 HClO4溶液中Pt、Pd、Pd4Ag中表面H吸附和内层H吸收(相变)的原位电输运测量
作者首先在经典强酸性溶液(HClO4)中研究了Pd、Pt、Au等材料在电催化过程中的表面H吸附及后续内层扩散(H吸收)导致的相变现象(统称为H Sorptions)。与Pt/Au相比,Pd在H还原电位区间经历了表面H吸附过程后,表面H原子持续向内层扩散并形成内层PdHx,导致原位电导率显著下降。这一现象可作为金属内层H吸收相变过程的特征信号,并可进一步通过ETS响应值和标定曲线得出氢化层中的H/M比例,实现电化学过程中针对金属H吸附/相变过程的原位定量表征和相关动力学测量。Pd4Ag合金在同样的还原电位区间存在类似相变过程,但原位氢化程度显著弱于Pd,体现出Ag掺杂导致了更弱的M−H相互作用。
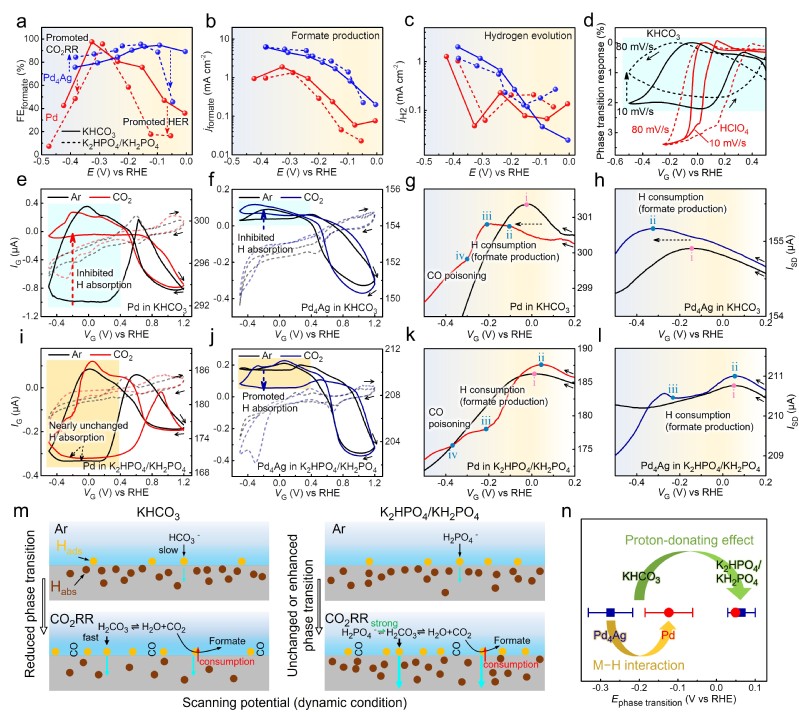
图3 Pd/Pd4Ag在CO2RR过程中的原位H吸附/相变研究
在此基础上,作者系统探究了不同条件下Pd催化二氧化碳还原反应中的原位H动力学和相变过程。首先,ETS信号明确展现出常用CO2RR缓冲电解质中更慢的H动力学特征。与空白背景相比,CO2RR反应过程中,CO2对于表面吸附H的消耗导致相变速度受到显著抑制,且这一特征信号与电解质中阴离子的给质子能力(pKa)高度相关。进一步实验结果表明,PBS比HCO3-更有利于维持界面处的H2CO3浓度和质子供应,加速H动力学过程。该结果展示出了电催化CO2RR表界面过程中阴离子、H2CO3和CO2分子在质子供给复杂局域化学平衡中的协同作用。更重要的是,ETS信号的特征细节(相变转折点、相变电位、相变程度等)可进一步体现掺杂Ag原子后,Pd4Ag与纯Pd相比更弱的CO毒化效应、M−H相互作用和H动力学。结合DFT计算结果进行进一步分析,这些结果来自于Ag对Pd的电子结构调控以及Ag对吸附物种(H*和CO*)的邻近效应影响,为合金类催化剂在CO2RR中的优异性能提供了合理的原位测量实验证据和原理解释。
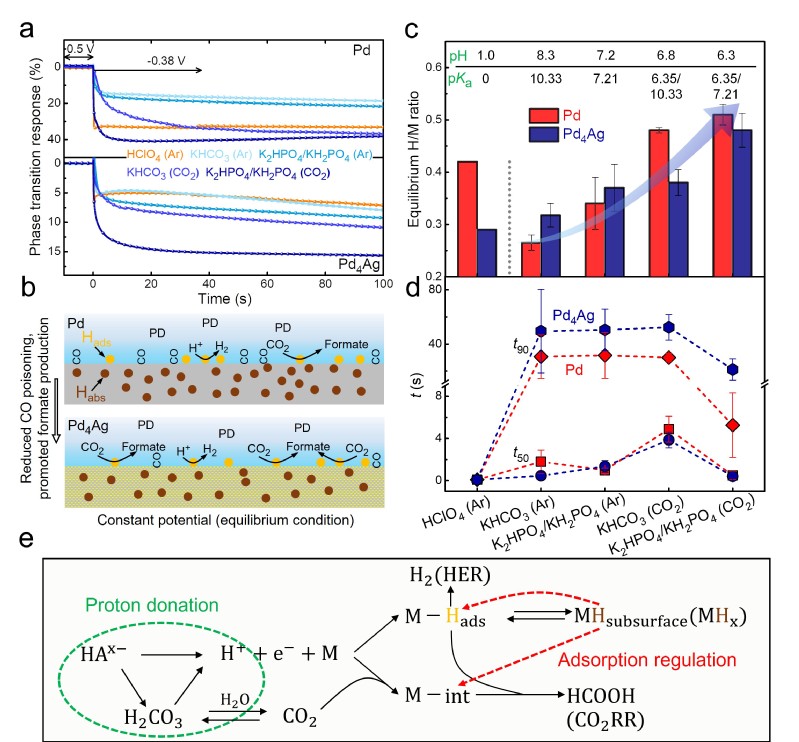
图4 恒电位条件下的H吸附/相变
由于催化剂的氢化程度将与催化过程中的活性位点(MHx)的组成直接相关,作者进一步研究了CO2RR工况条件(恒电位电解)下,表界面H动力学达到稳态后的近平衡M/H值及H动力学过程。由于更慢的H动力学,Pd在近中性条件下的相变程度较酸性条件下更低。更有意思的是,Pd4Ag在近中性条件下意外地体现出比酸性更高的相变程度。这一实验结果证实了酸性条件下Pd4Ag的表面H吸附能更强,抑制了其向体相扩散。该结论符合以往报道铂的酸碱H吸附和PdPt合金的H吸附/相变机制。此外,时间分辨的ETS进一步验证了K2HPO4/KH2PO4中,CO2的引入可以显著加速H动力学。所以,近中性条件下,电解质溶液(CO2产生的H2CO3和电解质离子)的给质子能力是影响表界面处复杂化学平衡网络的重要影响因素。在以往相关报道中,Pd氢化物的形成会造成钯金属d轨道中心的下移,有利于减弱CO毒化效应。本工作的结果显示在磷酸盐中,高氢动力学及相变程度的确有利于提升Pd4Ag在高过电位条件的催化选择性,对应于更高的甲酸法拉第效率和电流密度,这体现出氢动力学在影响氢化物活性位点形成中的重要作用。而纯钯由于较强的H吸附,相变难以进一步调控,H动力学的进一步加快则主要加速了竞争的析氢反应。
综上所述,本文采用片上电输运谱技术实现了钯基合金催化剂在二氧化碳电还原过程中的原位相变检测和工况电解条件下稳态H动力学的定量测量。该工作证明了在近中性条件下,电解质溶液的给质子能力(pKa)、界面处的CO2-H2CO3平衡及Pd表/内层的H吸附/吸收动力学,是决定CO2RR过程中氢化物(MHx)活性位点形成并调控反应中间体吸附的决定性影响因素。该工作进一步揭示了通过调控给质子过程可显著推动电催化表界面处MHx活性位点转变、中间体吸附、析氢和产甲酸的化学反应网络,这为优化电催化体系提供了新思路。
该研究工作得到了国家自然科学基金、中央高校科研经费、介观化学教育部重点实验室及生命分析化学国家重点实验室的支持。